BGI Genomics-led team leverages whole genome sequencing to release the first Chinese population blood atlas
2022-12-04
Whole genome sequencing technology can discover various known or unspecified viral sequences contained in human blood, which can provide an important database for viral infection prevention, vaccine development, viral genomic and epidemiological research. For example, numerous common cancers are associated with oncogenic viruses, including Epstein-Barr virus (EBV), hepatitis B and C viruses (HBV and HCV), and human papillomavirus (HPV).
In October 2022, BGI Genomics, Ruijin Hospital, Shanghai Jiao Tong University Institute of Translational Medicine leveraged the DNBSEQ sequencing platform to conduct an in-depth analysis of non-human genetic sequences in the whole genome sequencing (WGS) data of 10,585 people from China Metabolic Analysis Program (ChinaMAP) and constructed the first blood virological profile of the Chinese population. The results were published in the journal Cell Discovery and provide a reference for viral infection prevention and epidemiology.

This study established a WGS-based method to analyze viral sequences by extracting non-human gene sequences from WGS data of 10,585 individuals and identified 14 viruses that are widely present in the Chinese population, such as fingerprint virus, herpes B virus, human endogenous retrovirus, human adenovirus C, and hepatitis B virus.
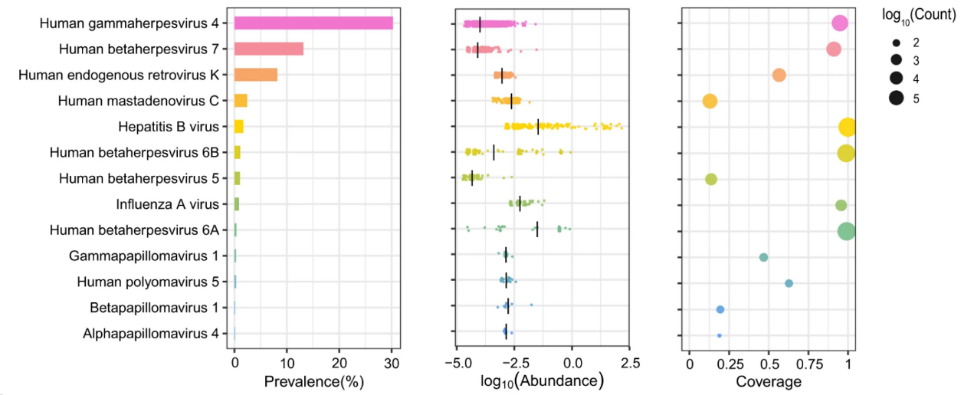
The highest detection rate was for Anellovirus, with fingerprint virus genetic sequences including TTV (Torque teno virus) and TLMV (TTV-like mini virus) found in 76.7% of individuals; HHV-4 (Human gammaherpesvirus 4, EBV) was detected in 30.3% of individuals, higher than that reported in the European population cohort (14%).
Herpesvirus B (Betaherpesvirus) was also widely detected, with HHV7 (Human herpesvirus 7), HHV6A (Human betaherpesvirus 6A), HHV6B (human betaherpesvirus 6B), and HHV5 (Human betaherpesvirus 5, HCMV) found in 13.2%, 0.36%, 1.09% and 1.03% of individuals, respectively.
Human endogenous retrovirus K (HERV-K), human mastadenovirus C (HMV) and hepatitis B virus (HBV) were found in respectively 8.20%, 2.41% and 1.69% of individuals.
In addition, the team detected HPV sequences in 50 individuals (0.47%), involving the subtypes Gammapapillomavirus 1, Betapapillomavirus 1 and Alphapapillomavirus 4
Most monitored virus – Hepatitis B virus
Hepatitis B is a major infectious disease in China and is still one of the major causes of liver cancer. The research team detected hepatitis B virus (HBV) sequences in 1.69% of individuals, consisting mainly of two subtypes (77%), HBV-B and HBV-C. Further, the team analyzed viral integration events in the human genome and found HBV-B viral sequence integration in 10 samples and HBV-C viral sequence integration in 18 samples, a result indicating that integration events were significantly associated with higher viral sequence abundance and that there were no significant enriched regions of HBV viral sequence integration sites on the human genome.
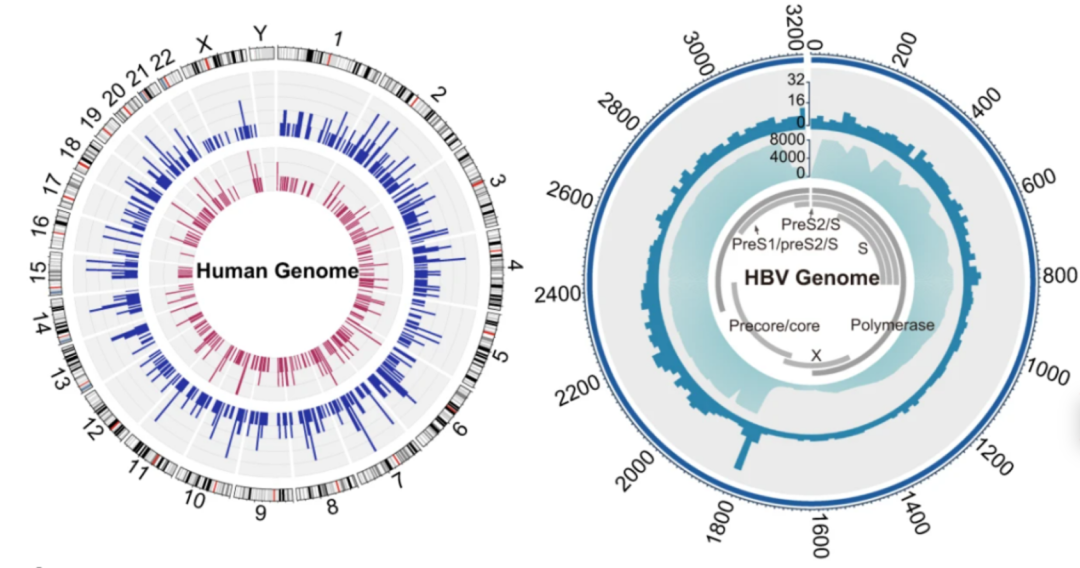
Left: integration sites of hepatitis B virus on the human genome; Right: integration sites of hepatitis B virus genome
The team further investigated whether there is a correlation between viral infection and genetic variation through genome-wide association studies (GWAS). The results showed that a missense mutation in the ACR gene was significantly associated with HHV6 viral carriage, and the locus was enriched in East Asian populations.
HHV6 belongs to the betaherpesviruses and includes two viruses with up to 90% genomic sequence identity, HHV6A and HHV6B. Although HHV6 infection is harmless to most people, studies have also found that HHV6 infection is associated with neurological diseases such as multiple sclerosis and Alzheimer's disease, indicating the potential harmfulness of HHV6 infection cannot be ignored. This insight provides an important database for future studies on the transmission mechanism of HHV6 virus, as well as genetic risk assessment of populations in regions with a high risk of HHV6 infection.
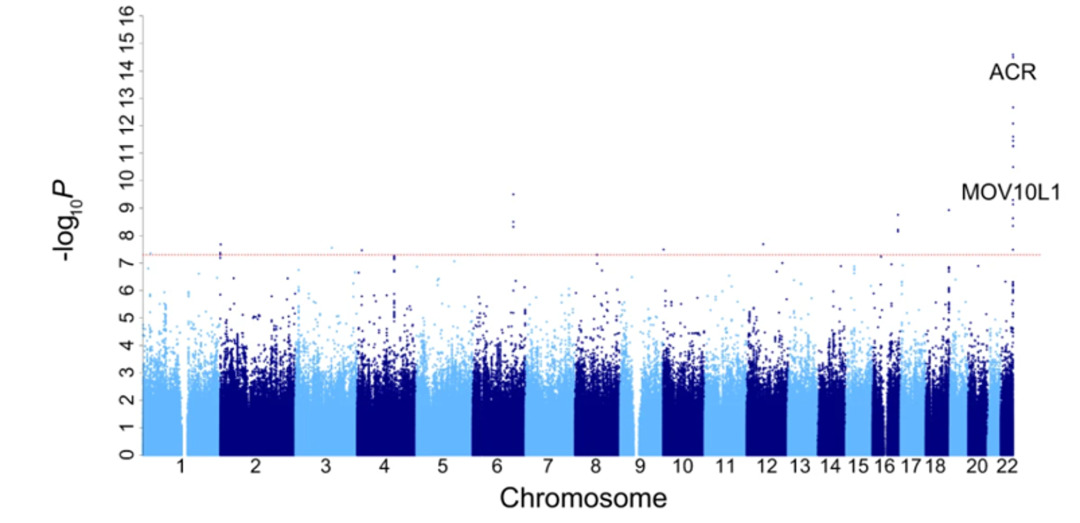
Genome-wide association analysis of HHV6 infection (Manhattan plot)
In summary, this study systematically investigated the blood viral group in a large-scale population using in-depth WGS data based on the DNBSEQ sequencing platform and analyzed the population carriage rate, viral abundance and geographic distribution of 14 viruses. It was found that EBV detected in 30% of individuals was the most frequent pathogenic virus carried in the population, HBV viral abundance in blood was associated with integration events, and the missense mutation in ACR was significantly associated with HHV6 virus carriage. These findings will provide important information for viral infection prevention and epidemiological studies. At the same time, they also provide references and lessons for larger population genomic projects.
This study is based on the China Metabolic Analysis Program (ChinaMAP). This study follows relevant regulations related to biological and medical research and has been approved by the BGI Genomics Ethics Committee and relevant ethics committees of research partners.
About BGI Genomics
BGI Genomics, headquartered in Shenzhen China, is the world’s leading integrated solutions provider of precision medicine. In July 2017, as a subsidiary of BGI Group, BGI Genomics (300676.SZ) was officially listed on the Shenzhen Stock Exchange.